Во месецот со најмалку дена во годината, во месецот кој пациентите со ретки болести го викаат „свој“, месецот кој е редок по своите ретки 28 дена и оваа година, четврта по ред, „Република“ медиумски ја поддржува камапњата „Ги запозаваме ретките болести“.
Шимке-имунокоскена дисплазија/Schimke-immuno-osseous dysplasia
МКБ 10: Q 77.7
Преваленца / распространетост: < 1/1.000.000
Шимке-имунокоскената дисплазија за првпат беше опишанa од д-р Р. Нил Шимке во 1971 година. Првично беше окарактеризиранa како нарушување со непропорционален низок раст, нефротски синдром, лимфопенија, неисправен клеточен имунитет. Болеста Шимке е многу ретка. Досега во литературата се пријавени околу 50 случаи, без очигледна предиспозиција за пол, етничко или географско потекло. Во Северна Америка, преваленцата на СИОД е проценета на 1 во 1-3 милиони живородени.
Симптоми:
Нискиот раст е резултат на сплескување на пршлените што доведува до скратување на вратот и трупот. Висината на возрасните е 136 – 157 см за мажи, и 98,5 – 143 см за жени. Бубрежната болест се манифестира со прогресивен кортикорезистентен нефротски синдром и често води кон бубрежно откажување и потреба од трансплантација. Обично, првите знаци на бубрежната болест се протеинурија и хипертензија.
Во имуниот систем дефектот е во целуларниот имунитет – нема доволно Т-клетки. Овие клетки се важни при идентификација на страните супстанции и при одбрана од инфекција. Како резултат на овој недостаток, зголемена е појавата на животозагрозувачки инфекции и болести со имунодизрегулација, како и автоимуни болести.
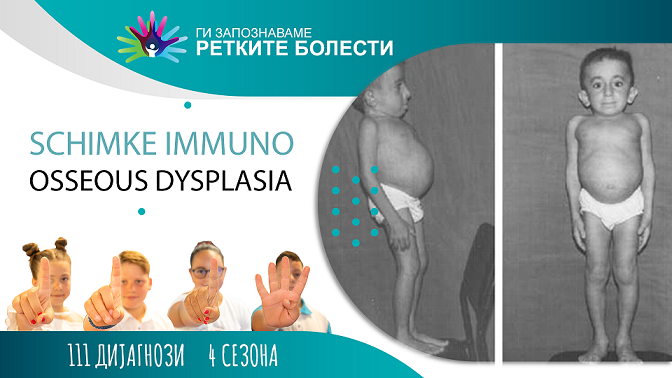
Како најчести манифестации се сретнуваат лордозата (изразена кривина во долниот дел на ‘рбетот), темни петна по кожата (хиперпигментации) кои се типични на градниот кош, напред или на грбот, широка база на носот и заоблен тип на нос. Поретко се сретнува акумулација на масни депозити или фиброзно ткиво во ѕидот на артериите (атеросклероза), намален крвоток во мозокот (церебрална ишемија), главоболки кои наликуваат на мигрена, намалена активност на тироидната жлезда (хипотироидизам), намален број на лимфоцити (лимфопенија), ентеропатија, нормоцитна или микроцитна анемија. неразвиеност на коските на карлицата (хипопластичен пелвис), микроцефалија и азоспермија кај машките, и ирегуларни менструации кај женските пациенти. Кај тешките случаи, симптомите на болеста се присутни уште од раѓање. Кај полесните, симптомите се јавуваат подоцна во детството.
Физички карактеристики:
Повеќето погодени лица имаат специфични физички карактеристики. Овие вклучуваат фина коса (60 %), тенка горна усна, широк низок мост на носот (68 %), луковичен врв на носот (83 %) и непропорционално низок раст (98 %). Дополнителните карактеристики вклучуваат прекумерно искривување на лумбалниот ʼрбет навнатре (лумбална лордоза, 84 %), испакнат стомак и хиперпигментирани макули (85 %) на трупот и повремено на вратот, лицето, рацете и нозете.
Дијагноза:
Дијагнозата се заснова на внимателно клиничко, биохемиско и радиолошко оценување со остеопенија, овоидни и сплескани вертебрални тела, и хипопластични феморални глави и ацетабуларни покриви како типични радиографски карактеристики. За да се потврди дијагнозата може да се направи молекуларно тестирање.
Етиологија:
Болеста на Шимке е предизвикана од мутации во СМАРКАЛ-1 генот кој го енкодира протеинот хХАРП одговорен за ремоделирање на хроматинот во хромозомите.
Наследување:
Мутациите СМАРКАЛ-1 генот се наследуваат автозомно рецесивно, што значи дека ризикот за болест е поголем ако се присутни мутации во двете копии на СМАРКАЛ-1 генот во секоја клетка. Родителите на лицата кои боледуваат од автозомно рецесивна болест, секој носи една копија мутиран ген, но тие типично не покажуваат знаци на болеста. Соодветно е да се понуди генетско советување (вклучително и дискусија за потенцијалните ризици за потомството и опциите за репродукција) на млади возрасни лица кои се носители или имаат ризик да бидат носители. Пренаталната дијагноза е можна кога патогената варијанта е претходно идентификувана кај член на семејство.
Третман:
Бидејќи болеста зафаќа повеќе органски системи, водењето на пациентите треба да е мултидисциплинарно. За пациентите со терминална бубрежна инсуфициенција, терапија на избор е бубрежната трансплантација. Заради постоечкиот имун дефицит, имуносупресивната терапија по бубрежната трансплантација е асоцирана со зголемен ризик од отфрлање и тешки опортунистички инфекции. За имунодефициенцијата и другите хематолошки абнормалности, терапија на избор е трансплантација на коскената срцевина.
Прогноза:
Повеќето пациенти фатално завршуваат во детската возраст или во раната адолесценција. Пациентите со поблага форма и доцен почеток, можат да поживеат до адултната возраст. Кај повеќето пациенти очекуваниот животен век е ограничен на детство или рана адолесценција, заради мозочен удар, инфекции, откажување на коскената срж и ренална инсуфиенција. Забележано е преживување до зрелост, кај пациенти со поблага и подоцнежна форма на болеста и успешно управување со бубрежните манифестации.
за проектот:
Гордана Лолеска, Супер радио и Здружението „Живот со предизвици“ и овој февруари, во рамките на четвртата сезона од проектот „Ги запознаваме ретките болести“, Ве запознаваат со ретките болести во Република Македонија, со надеж дека преку овие нови 28 ретки дијагнози сите заедно ќе успеене да го кренеме гласот за проблемите со кои се среќаваат лицата со ретки болести, но и нивните семејства.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29-ти февруари, „редок“ датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има „редок“ број на денови.
Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008-ма година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година. Република Македонија за прв пат се вклучи во одбележување на овој редок ден во 2013-та година.
Republika.mk - содржините, графичките и техничките решенија се заштитени со издавачки и авторски права (copyright). Крадењето на авторски текстови е казниво со закон. Дозволено е делумно превземање на авторски содржини (текст и фотографии) со ставање хиперлинк до содржината што се цитира.





Comments are closed for this post.