Во месецот со најмалку дена во годината, во месецот кој пациентите со ретки болести го викаат „свој“, месецот кој е редок по своите ретки 28 дена и оваа година, четврта по ред, „Република“ медиумски ја поддржува кампањата „Ги запознаваме ретките болести“.
Мелкерсон-Розенталов синдром/Melkersson-Rosenthal syndrome
МКБ: G 51.2
Преваленца/распространетост: непозната
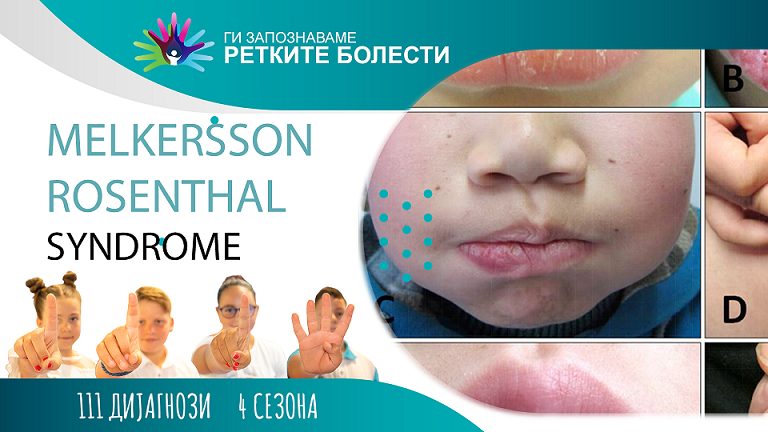
Мелкерсон-Розенталов синдром (МРС) е ретко невролошко нарушување кое се карактеризира со тријада составена од рекруентни симптоми, и тоа: долготраен орофацијален едем, особено на едната или двете усни (грануломатозен хиелитис), рекруентна лицева парализа и набразден јазик.
Синдромот за првпат е опишан во 1920 година од Ернест Густав Мелкерсон кај 35-годишна пациентка со орофацијален едем и парализа на фацијалниот нерв. Поврзаноста на парализата на фацијалниот нерв и набраздениот јазик е опишана во 1931 година од Розентал.
Почетните симптоми се јавуваат помеѓу 25-та и 40-та година од животот. Жените имаат поголема предиспозиција во однос на мажите (2:1), додека пак, МРС е редок кај децата. Опишани се досега 30 случаи кај деца, со тоа што почетните симптоми се јавуваат помеѓу 7-та и 12-та година.
Етиологија:
Етиологијата на Мелкерсон-Розенталовиот синдром е недоволно разјаснета. Етиопатогенезата на ретките педијатриски случаи е поврзана со инфекцијата предизвикана од Херпес Симплекс вирусот, хередитарни грануломатозни нарушувања, туберкулоза и лепра, хронични инфекции, Даунов синдром, псоријаза, тироидитис, мултиплекс склероза, кератитис, Вегнерова грануломатоза, дијабет, саркоидоза, улцеративен колитис и алергии. Респираторните инфекции од инфлуенца-вирусот можат да бидат тригер-фактор за ремисија на болеста.
Постојат докази кои сугерираат дека Мелкерсон-Розенталовиот синдром има наследна компонента бидејќи забележано е фамилијарно наследување кај неколку пациенти. Сепак, клиничката презентација варира и го прави процесот на дијагностицирање еден вид предизвик за клиничарите.
Симптоми:
Мелкерсон-Розенталовиот синдром обично се дефинира со класичната тријада од симптоми (лицева парализа, повторувачки орофацијален едем и набразден јазик). Неговата комплетна клиничка презентација е невообичаена. Опишана е во само 10-25 % од дијагностицираните случаи.
Парализа на лице:
Парализата на лице има невролошко потекло и се карактеризира со неподвижност на мускулите кои се надразнети од лицевиот/фацијален нерв. Оваа состојба најчесто се јавува како резултат на привремено или трајно оштетување на фацијалниот нерв предизвикано од воспаление, оштетување на ткивата, и сл. Фацијалниот нерв е одговорен за инервација на различни подрачја на лицето и околните структури. Неговата функција е контрола врз мимиката на лицето која овозможува изразување емоции, артикулација на гласовите, звуци, јадење, итн. Присуството на разни патолошки процеси можат да предизвикаат слабеење или парализа на подрачјето кое е инервирано од фацијалниот нерв. Кај Мелкерсон-Розенталовиот синдром, парализата на лицето може да има периферен карактер со зафаќање на едната страна на лицето, со повторувачки карактер. Овој симптом се јавува кај повеќе од 30 % од заболените. Обично е со брз развој, за време од околу 24-48 часа.
Орофацијален едем:
Орофацијалниот едем е присутен кај 80 % од случаите. Се карактеризира со присуство на абнормална и патолошка акумулација на течности која создава воспаление или оток на зафатеното подрачје. Може делумно или во потполност да го зафати лицето, јазикот, непцата или оралната слузница. Најчесто се зафатени усните, посебно горните (грануломатозен хиелитис). Орофацијалниот едем често е пропратен со фебрилни епизоди и други симптоми. Овој клинички симптом обично се јавува во рок од неколку часа со тенденција за кратко време да добие повторувачки тек.
Набразден јазик:
Овој вообичаен симптом се карактеризира со надолжна вдлабнатина на средината и со појава на попречни пукнатини. Овој симптом е поврзан со генетските абнормалности и обично е пропратен со намалено чувство за вкус и парестезии.
Дијагноза:
Дијагнозата на овој синдром се поставувa врз основа на сомнежот кон класичната симптоматска тријада. Не постои лабораториски тест кој може да го утврди неговото присуство. Хистопатолошките испитувања често се користат при анализа на едемот.
Третман:
Не постои специфичен третман за Мелкерсон-Розенталовиот синдром. Кортикостероидите се основен елемент во третманот на овој синдром кои овозможуваат подобрување кај 50-80 % од пациентите и ги редуцираат шансите за ремисија за 60-75 %. Во третманот се вклучени и нестероидните антиинфламаторни лекови, антихистаминици и антибиотици. Кај некои пациенти се препорачува и употреба на имуносупресиви. Хируршкиот третман и радијацијата се препорачуваат за редукција на абнормално отечените усни.
за проектот:
Гордана Лолеска, Супер радио и Здружението „Живот со предизвици“ и овој февруари, во рамките на четвртата сезона од проектот „Ги запознаваме ретките болести“, ја запознаваат јавноста со ретките болести во Република Македонија, со надеж дека преку овие нови 28 ретки дијагнози сите заедно ќе успеат да го кренат гласот за проблемите со кои се среќаваат лицата со ретки болести, но и нивните семејства.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29-ти февруари, „редок“ датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има „редок“ број на денови.
Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008-ма година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година. Република Македонија за прв пат се вклучи во одбележување на овиј редок ден во 2013-та година.
Republika.mk - содржините, графичките и техничките решенија се заштитени со издавачки и авторски права (copyright). Крадењето на авторски текстови е казниво со закон. Дозволено е делумно превземање на авторски содржини (текст и фотографии) со ставање хиперлинк до содржината што се цитира.









Comments are closed for this post.