Во месецот со најмалку дена во годината, во месецот кој пациентите со ретки болести го викаат „свој“, месецот кој е редок по своите ретки 28 дена и оваа година, четврта по ред, „Република“ медиумски ја поддржува камапањата „Ги запознаваме ретките болести“.
Фамилна хипомагнеземија, хиперкалциурија и нефркалциноза/Familial hypomagnesemiа, hypercalciuria and nephrocalcinosis
MKБ: E 83.4
Преваленца/распространетост: 1/1.000.000
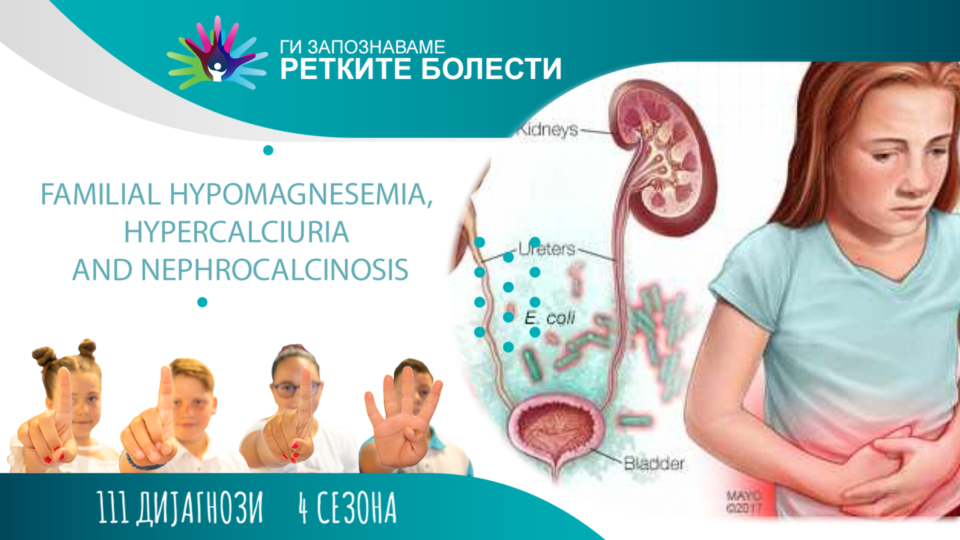
Фамилна хипомагнеземија, ниско ниво на магнезиум во крвта, која е проследена со хиперкалциурија, зголемено излачување на калциум преку урината, и нефрокалциноза, натрупување на калциум во бубрежното ткиво, е ретка автозомно рецесивна болест која се карактеризира со губење на магнезиум и калциум преку бубрезите, развој на нефрокалциноза и бубрежни камчиња во раното детство. Често е асоцирана со прогресивно губење на бубрежната функција, прогресивна бубрежна слабост во детството или адолесценцијата. Кај пациените се јавуваат чести уринарни инфекции, зголемено излачување на урина и тоа во количини повеќе од 3 литри за 24 часа, зголемено чувство за жед пропратено со зголемено внесување течности, доцнење во раст и развој, болки во абдомен, губиток на вид, неконтролирано движење на очите – нистагмус, и останати офталмолошки симптоми. Поставувањето на дијагноза е обично помеѓу 1-8 години, со тоа што почесто се јавува кај женските отколку кај машките деца. Фамилната хипомагнеземија првпат е опишана во 1969 година, но соодветно е класицифицирана во 1987 година. До денес, во литературата се пријавени приближно 200 случаи.
Причини:
Фамилната хипомагнеземија со хиперкалциурија и нефрокалциноза е автозомно рецесивна болест која е предизвикана од мутација во генот што го кодира протеинот клаудин-16 и генот што го кодира протеинот клаудин-19. Во зависност од тоа во кој од двата гена е мутацијата, ќе зависат и симптомите кај пациентите, со што кај оваа болест се опишуваат два поттипа, и тоа фамилна хипомагнеземија со хиперкалциурија и нефрокалциноза, и фамилна хипомагнеземија со хиперкалциурија и нефрокалциноза со окуларно засегање. Двата протеина се наоѓаат во асцедентниот крак на Хенлеовата петелка, при што имаат улога во реапсорпцијата на магнезиум и калциум. Покрај Хенлеовата петелка, клаудин-19 протеинот се наоѓа и во пигментниот епител на очната мрежница, па од таму и произлегуваат симптомите поврзани со очите и видот кај овие пациенти.
Симптоми:
Кај овие пациенти уште од раното детство се појавуваат чести уринарни инфекции. Покрај нив, најчести симптоми се наодот на нефрокалциноза, бубрежните камчиња, зголеменото излачување на урина, зголемено чувство за жед и внес на течности, заостанување во раст и развој, како и болки во абдоменот. Исто така, кај некои од пациентите се опишани и грчеви и болки во мускулите поради намаленото ниво на магнезиум. Со текот на времето, најчесто во адолесценцијата, кај некои случаи и подоцна во третата деценија од животот, оваа болест кулминира со прогресивно губење на бубрежната функција. Во потешките случаи кај пациентите со мутација на клаудин-19 протеинот се јавува макуларен колобом, нистагмус и губиток на вид.
Дијагноза:
Дијагнозата на оваа болест се засновува на тријасот на симптоми: хипомагнеземија, хиперкалциурија и нефрокалциноза. Ултразвучниот наод на уринарниот тракт ни покажува таложење на калциум во бубрежното ткиво. Намалениот калциум и зголемената мочна киселина во крвта се само придружни наоди. Кај оваа болест нивото на паратироидниот хормон е висок пред појавата на хронична бубрежна болест. Очните промени се детектираат со помош на детален офталмолошки преглед, т.е. со преглед на очното дно. Но, сепак дефинитивна дијагноза се поставува со потврда од страна на генетски скрининг за мутација на гените клаудин-16 и клаудин-19. Наследувањето е автозомно рецесивно. Треба да им биде понудена генетска консултација на паровите кои се со ризик (двете индивидуи се носители на мутацијата која ја предизвикува оваа болест) за да се информираат дека постои 25 % можност да се роди дете кое ќе биде заболено. Диференцијалната дијагноза ги вклучува Бартеровиот синдром, автозомната доминантна хипокалцемија, Дентовата болест, дистално-реналната тубуларна ацидоза, наследниот хипофосфатемичен рахит со хиперкалциурија, како и тубуларното нарушување, кои севкупно предизвикуваат рана нефрокалциноза.
Последици:
Последици од фамилната хипомагнеземија со хиперкалциурија и нефрокалциноза е терминалната фаза на бубрежна болест кај 50 % од пациените која се јавува до 20-годишна возраст. Пациените со мутација на клаудин-19 протеинот, имаат поголем ризик од развивање на хронична бубрежна болест.
Третман:
Третманот вклучува давање магнезиум-суплементи, како и тиазидни диуретици за да се намали уринарното излачување на калциум и прогресијата на нефрокалцинозата. Терапијата е со цел да се одложи прогресијата на хроничната бубрежна болест. Бубрежната трансплантација е оптимален третман кај ова заболување. Кај оние пациенти кои страдаат од сериозни очни воспаленија, се препорачува и имплантација на леќа во окото.
за проектот:
Гордана Лолеска, Супер радио и Здружението „Живот со предизвици“ и овој февруари, во рамките на четвртата сезона од проектот „Ги запознаваме ретките болести“, Ве запознаваат со ретките болести во Република Македонија, со надеж дека преку овие нови 28 ретки дијагнози сите заедно ќе успеене да го кренеме гласот за проблемите со кои се среќаваат лицата со ретки болести, но и нивните семејства.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29-ти февруари, „редок“ датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има „редок“ број на денови.
Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008-ма година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година. Република Македонија за прв пат се вклучи во одбележување на овој редок ден во 2013-та година.
Republika.mk - содржините, графичките и техничките решенија се заштитени со издавачки и авторски права (copyright). Крадењето на авторски текстови е казниво со закон. Дозволено е делумно превземање на авторски содржини (текст и фотографии) со ставање хиперлинк до содржината што се цитира.








Comments are closed for this post.